

PURA Syndrome Cure Roadmap
PURA Syndrome Cure Roadmap
Prepared for Jack’s Tomorrow by Perlara PBC
“I have reviewed the proposed program by Perlara to develop treatments for children with Pur alpha syndrome. I was impressed by the thorough consideration of the literature and of possible mechanisms for moving forward. I strongly agree with the proposed mechanisms for moving forward and their prioritization.”
-Professor Andrew Bergemann, PhD (cloned the PURA gene in 1992)
“
Watch Our 2022 PURA Syndrome Research Update
Vision
Towards a future where people with PURA syndrome will lead healthy, interactive, and fulfilled lives. This roadmap describes the most promising avenues for finding a treatment for patients with PURA Syndrome. Based on the current knowledge about the gene and how it functions, we recommend moving forward with three therapeutic strategies: antisense oligonucleotides (ASOs), drug repurposing and gene replacement. In parallel, the document further discusses other therapeutic modalities. We explore the preclinical steps to be undertaken and encourage the establishment of a cure network/team dedicated to accelerating clinical therapeutic development and improving lives of patients with PURA-related disorders.
Executive Summary
The PURA Syndrome Cure Roadmap aims to stimulate the development of novel therapeutics for PURA syndrome, a rare neurodevelopmental disorder caused by heterozygous dominant variants in the PURA gene. This Roadmap was developed and revised following dozens of interviews and a thorough review process involving academic scientists, clinicians and biotech (see authors’ note).
Key Recommendations
After an extensive interview process with many experts, our top recommendations for areas to fund through Jack’s Tomorrow in order to find a cure for PURA Syndrome include both basic science and therapeutic tracks:
Basic Science 1: Develop an in vitro model for PURA syndrome by investing in efforts to develop patient-derived iPSCs and further differentiate them into neuronal and skeletal muscle cell types. Characterize variants and mechanisms of disease.
Basic Science 2: Identify screenable phenotypes in cell culture for the development of assays – stress granules dynamics and critical targets of PURA.
Basic Science 3: Establish in vivo new mouse models for PURA syndrome – conditional knockout (available from a repository) and patient avatars.
Basic Science 4: Generate in vivo fruit fly and worm models for PURA syndrome – patient avatars, and further characterize the existing knockout and knockdown models; perform genetic screens.
Therapeutic Track 1: ASOs– design allele-specific and allele-agnostic ASOs to knockdown PURA variants and/or upregulate the healthy PURA allele (based on the outcome of the variant characterization); test these in the in vitro and in vivo models.
Therapeutic Track 2: Drug repurposing – screen for small molecules that alleviate phenotypes in vivo in fruit flies and worms; then test lead candidates in cell culture and mouse models.
Therapeutic Track 3: Gene replacement – perform gene replacement proof of concept studies in the newly established mouse models via AAV9.
Basic Science Tracks: The efforts around the basic science are to further our understanding of the effect of different pathogenic PURA variants and identify the mechanism(s) of action and regulation of PURA.
Variants
First, it is imperative to characterize the variants by studying the differences between the healthy vs mutated versions of the gene. While over 90 pathogenic variants have been reported - including deletion, nonsense, missense, and frameshift variants - no functional studies have been performed to clarify how these variants cause the disease. We plan to develop both in vitro (outside an organism) and in vivo (in an organism) models to answer this and other unsolved questions in the PURA syndrome field. Based on current knowledge of PURA biology and patients’ clinical features, neuronal and skeletal muscle cells are likely the most relevant cell types in PURA syndrome. Current technology allows us to convert Jack’s fibroblasts or peripheral blood mononuclear cells (PBMCs) into induced pluripotent stem cells (iPSCs) and then differentiate them into various types of neurons or into skeletal muscle cells, providing an in vitro model for functional testing and screening of successful therapeutics.
Mouse Model
To study the disease within a living organism, or “in vivo”, two Pura knockout (deletion) mouse models were previously generated by two independent groups and shown to recapitulate the main clinical phenotypes of PURA patients. However, both mouse models have limitations dictated by the strategy used for their generation, and partially contradicting observations were reported on the two mouse models. We plan to generate a new knockout mouse model as well as mice that carry patients’ variants, taking advantage of the latest, most advanced genome editing tools.
Additional Animal Models
We will also use Drosophila melanogaster (flies) and Caenorhabditis elegans (worms) as model organisms for PURA loss of function studies, generation of patient avatars and drug repurposing screening.
Detailed molecular and phenotypic characterization of these in vitro and in vivo models will allow us to gain more insight into the pathobiology of PURA syndrome, and to find measurable outcomes for the development of therapeutics.

Finding a Treatment
Following the extensive review of the PURA literature, we plan to prioritize three main therapeutic paths, including the use of ASOs, drug repurposing, and a gene replacement strategy.
ASO - ASOs are small nucleic acid-based sequences that can target specific RNA sequences. Based on the knowledge acquired from the basic science track, we will design either variant specific ASOs to prevent expression of the mutant allele and/or ASOs targeting various regulatory mechanisms of PURA gene expression to upregulate the healthy allele. The goal is to rid the cell of mutated PURA mRNA (if needed), and to provide a functional level of healthy PURA mRNA. We will test the various ASOs in the in vitro (cell lines) and in vivo models to assess for alleviation of symptoms, toxicity, and optimal dosing.
Drug Repurposing - In parallel, we will screen a library of small molecules to determine whether any of these drugs can be repurposed for PURA syndrome patients. We will identify if any of the thousands of FDA-approved drugs in the library can lessen symptoms in specific cell lines or in in vivo models. Potentially, this will generate drug candidates that could be further tested through clinical trials.
Gene Replacement - The third recommended path is the development of a gene therapy strategy to deliver a replacement healthy copy of the PURA gene to disease target tissues via non-integrating adeno-associated viral vectors (AAVs).
Authors
Barbara Celona, PhD (Cure Guide at Perlara PBC)
Carole Shum, PhD (Cure Guide at Perlara PBC)
Ethan O. Perlstein, PhD (CEO of Perlara PBC & Maggie’s Pearl LLC)
Authors’ note
This PURA Cure Roadmap was prepared between April 2022 and June 2022. After reviewing the PURA primary literature, Perlara conducted interviews with over a dozen key opinion leaders. Following those discussions and research, the Perlara team drafted the PURA Syndrome Cure Roadmap and invited opinion leaders and additional reviewers from academia, medicine, and industry to participate in the review process and share their feedback.
The following reviewers provided feedback on the draft and the Roadmap (September 2022 version v2) was finalized on September 14, 2022.
The PURA Cure Roadmap was presented to families on October 24, 2022 in a Zoom community briefing. Video is available here.
Reviewers
Alexandra Amen, PhD
Director, Asset Acquisition
BridgeBio
Andrew D. Bergemann, PhD
Senior Faculty
Baylor College of Medicine
Allyson Berent, DVM, Dipl. ACVIM
CSO, Foundation for Angelman Syndrome Therapeutics
COO, GeneTx Biotherapeutics
Animal Medical Center, Director Interventional Endoscopy Service
Gianluca Cestra, PhD
Principal Investigator
Institute of Molecular Biology and Pathology (IBPM), National Research Council (CNR)
c/o University of Rome La Sapienza, Dept. of Biology and Biotechnology Charles Darwin
Lishan Chen, PhD
Senior Scientist
Mahzi Therapeutics
Wendy Chung, MD, PhD
Kennedy Family Professor of Pediatrics and Medicine
Departments of Pediatrics & Medicine
Columbia University
Jessica Duis, MD
Associate Professor of Pediatrics & Genetics
Children’s Hospital Colorado and University of Colorado
Andrea Foote, PhD candidate
Departments of Medicine & Biochemistry
University of Vermont College of Medicine
Vincenzo A. Gennarino, PhD
Assistant Professor
Department of Genetics & Development
Columbia University Irving Medical Center
Zachary Grinspan, MD, MS
Associate Professor of Healthcare Policy & Research and Pediatrics
Director of the Pediatric Epilepsy Program
Weill Cornell Medicine
Robert J. Kelm, Jr., Ph.D.
Associate Professor
Departments of Medicine & Biochemistry
University of Vermont College of Medicine
Eric Marsh, MD, PhD
Associate Professor of Neurology at the Children’s Hospital of Philadelphia
Clinical Director, Orphan Disease Center
Department of Neurology, Perelman School of Medicine
University of Pennsylvania
Dominique C. Pichard, MD
Chief Science Officer
International Rett Syndrome Foundation
Devorah Segal, MD, PhD
Clinical Assistant Professor of Neurology and Pediatrics
Associate Director, Comprehensive Neurofibromatosis Center
Department of Neurology, Division of Child Neurology
NYU Langone Medical Center
Victoria Suslovitch, MS, CGC
Genetic Counselor
Harvard Medical School
Kimara L. Targoff, MD
Associate Professor
Division of Cardiology
Department of Pediatrics
Columbia University
Yael Weiss, PhD
CEO
Mahzi Therapeutics
Timothy Yu, MD, PhD
Associate Professor
Harvard Medical School
We would like to thank these opinion leaders and reviewers for their insights, thoughtful comments, and efforts toward improving our PURA Syndrome Cure Roadmap. In the following summary, we highlight recommendations and comments from our reviewers.
Feedback
Our reviewers stressed that clarifying the mechanism of disease in PURA syndrome is critical for the prioritization of the proposed basic science and therapeutic tracks.
There was consensus regarding the need to determine if the dominant phenotype of the heterozygous PURA variants is due to haploinsufficiency and loss of function, or gain of function, dominant negative effects, or a combination of the above. Most reviewers also agreed with the need to address additional basic science knowledge gaps such as PURA critical functions and downstream targets, disease target cell types, gene regulatory mechanisms, dosage sensitivity, phenotype reversibility, and therapeutic window of intervention.
For the basic science track, many reviewers highlighted the importance of establishing in vitro and in vivo PURA knockout models as well as patient avatar models to determine the mechanism of disease, gain insight into the pathobiology of PURA syndrome and identify measurable robust outcomes for early phase drug development and preclinical testing.
In particular, multiple reviewers advocated for the generation of knockout and patient iPSCs cell lines, differentiation into neuronal and skeletal muscle cells and testing of cell sensitivity to stress granule promoting agents. In addition, several reviewers recommended moving forward with genetic screens in Drosophila melanogaster and Caenorhabditis elegans to quickly improve our understanding of PURA function, and for using these model organisms to quickly generate patient avatars and perform drug repurposing screens. The importance of a reliable knockout model in mice was also emphasized, along with the need to investigate the robustness of the phenotypes previously described in the literature and the utility of the mouse model for ASO and gene replacement proof of concept studies.
For the therapeutics tracks, our original Roadmap aimed to prioritize ASOs and drug repurposing as the two initial main therapeutic tracks. While most reviewers agreed that ASOs are a short-term safe option, some of them raised concerns about redosing and the challenge that long-term repeated administration constitutes for the patients. In addition, as some of the reviewers pointed out, given the lack of mutational hotspots and the identification of over 90 pathogenic variants of PURA spread across the entire sequence, gene replacement would have the advantage to potentially cure all patients independently by the type and location of the variant if pure haploinsufficiency is the mechanism of disease.
Based on this feedback and in light of additional information that came to our attention during the review process - specifically, the availability of a new conditional knockout mouse that could be used for proof of concept studies - we have now included gene replacement as therapeutic track to be initiated in Year 1.
Regarding the drug repurposing path, we are thankful to the many reviewers who recommended the inclusion of drugs in use for relevant disorders that have symptoms overlapping with PURA syndrome. We have included their recommendations in this revised version of the Roadmap.
Finally, we would like to thank Dr. Andrew Bergemann, who first identified and characterized PURA in 1992, for his critical review of the Roadmap and the kind words of support quoted below.
“I have reviewed the proposed program by Perlara to develop treatments for children with Pur alpha syndrome. I was impressed by the thorough consideration of the literature and of possible mechanisms for moving forward. I strongly agree with the proposed mechanisms for moving forward and their prioritization.
In terms of prioritization and emphasis, I would argue for genetic screens looking for mutations that complement or exacerbate the phenotypes of the existing Pur alpha models in Caenorhabditis elegans. At this moment, the lack of clearly defined developmental functions for Pur alpha remains a challenge to progress. While proteomic approaches identifying the Pur alpha interactome would unquestionably be helpful in this regard, the genetic screens have the potential to very quickly add to our understanding of function, and subsequently the generated models will be useful for early phase drug development and testing.
One last thought is that the current state of the Pur alpha field, with both clinical significance and some very suggestive basic Science results, should allow for the recruitment of more researchers into the field, including some of the leaders in the field of developmental neurobiology”.
Roadmap updates
This Roadmap is considered version 2 (v2) and it will require quarterly comprehensive reviews to incorporate progress reports and course corrections.

Introduction
PURA syndrome is a rare autosomal dominant neurodevelopmental disorder caused by genetic alterations in the Purine-rich element-binding protein A (PURA) gene, located on chromosome 5q31 (Hunt et al., 2014; Lalani et al., 2014; Tanaka et al., 2015). To date, about 500 affected individuals have been identified (source: PURA Syndrome Foundation). Most patients present with moderate to severe neurodevelopmental delay, absence of speech and lack of independent ambulation. Early-onset problems include severe neonatal hypotonia, excessive hiccups, recurrent apneas, hypersomnolence, hypothermia, feeding difficulties, seizures, and abnormal nonepileptic movements. Other less common manifestations comprise congenital heart defects, urogenital malformations, and various skeletal, ophthalmological, gastrointestinal, and endocrine anomalies (Reijnders et al., 2018).
The diagnosis of a PURA-related neurodevelopmental disorder is established in a proband with either a heterozygous PURA pathogenic variant (90% of affected individuals) or a microdeletion of 5q31.3 that encompasses all or part of PURA (10%). Almost all pathogenic sequence variants are de novo and to date, all reported 5q31.3 deletions have been de novo. Individuals with intellectual disability and 5q31.3 deletion were first identified in 2011 (Shimojima et al., 2011; Hosoki et al., 2012). In these individuals the size of the deletion can range from several thousand to several million base pairs and can include the genes Neuregulin2 and PURA (Hosoki et al., 2012). In 2014, heterozygous de novo mutations in the PURA gene were identified by whole exome sequencing (WES) in patients with symptoms similar to 5q31.3 microdeletion syndrome, suggesting that haploinsufficiency of PURA accounts for many of the clinical features in these patients (Hunt et al., 2014).

PURA (GRCh38/hg38, chr5:140,114,109-140,125,619), is a single-exon gene that encodes Pur-α, a highly conserved and ubiquitously expressed multifunctional protein of 322 amino acids (AA) in length (Bergemann et al., 1992a). Pur-α is a member of the Pur family of nucleic acid binding proteins which also includes Pur-β (Kelm et al., 1997) and two forms of Pur-γ (Liu et al., 2002). Pur-α consists of an N-terminal glycine-rich domain (AA 7–53), three central conserved repeats, Pur repeat I-II-III (AA59-123; 142-209; 218-278), and a C-terminal Gln/Glu-rich domain (AA282–322) (Graebsch et al., 2009; Weber et al., 2016). Pur repeats I and II fold into an intramolecular dimer that serves as a sequence-specific DNA/RNA-binding domain. The third repeat has been suggested to mediate intermolecular homo- and hetero-dimerization (Weber et al., 2016).
Pur-α binds single-stranded and double-stranded DNA and RNA sequences with preference for single-stranded GGN-repeats (Bergemann et al., 1992b; Gallia et al., 2000; Graebsch et al., 2009; Johnson et al., 2013) and has helix-unwinding capability (Weber et al., 2016). Pur-α has been implicated in a variety of cellular processes including DNA replication and cell cycle progression, DNA repair, transcription, RNA transport, translation, and membraneless organelle dynamics (for a review: Molitor et al., 2021; Daniel et al., 2018).

Pur-α is essential for normal postnatal brain development as it is involved in neuronal proliferation, dendrite maturation, mRNA transport to translation sites in hippocampal neurons, and formation and maturation of myelin (Hokkanen et al., 2012; Johnson et al., 2006, 2013; Khalili et al., 2003; White et al., 2009). Two independently generated knockout mouse models appear normal at birth but develop neurologic features, including ataxic gait, hind limb weakness, and continuous and increasingly severe tremors (Khalili et al., 2003; Hokkanen et al., 2012). Expression of Pur-α in the mouse brain is developmentally regulated and peaks at 15 to 18 days after birth, approximately the time the Pura knockout mice die (Khalili et al., 2003). Heterozygous mice display a less severe phenotype with still a significant reduction in number of neurons in the cerebellum and hippocampus, decreased tone, gait abnormalities, memory deficit and a higher prevalence of spontaneous early deaths (Barbe et al., 2016). In mice, Pur-α is detected predominantly in the cytoplasm, stress granules and neuronal transport granules.
Beyond PURA syndrome, PURA has been implicated in the pathogenesis of Fragile X-associated tremor/ataxia syndrome (FXTAS), Fragile X syndrome (FXS) and Amyotrophic Lateral Sclerosis (ALS). Pur-α colocalizes with the fragile X mental retardation protein FMRP (encoded by FMR1) in rat hippocampal neurons at dendritic junctional translation sites (Johnson et al., 2006), and interacts with rCGG repeats in the FMR1 gene, which is overexpressed in patients with FXTAS and silenced in individuals with FXS (Jin et al., 2007). Interestingly, ectopic expression of Pur-α suppresses neurodegeneration in a fly model of FXTAS (Jin et al., 2007) and mutations at key amino acid residues involved in DNA/RNA-binding abolish this neuroprotective effect (Weber et al., 2016). Similarly, Pur-α binds to expanded rGGGGCC repeats (Cooper-Knock et al., 2014; Rossi et al., 2015) and its ectopic expression suppresses C9orf72 ALS-related neurodegeneration in flies (Xu et al., 2013) and zebrafish (Swinnen et al., 2018). However, the exact mechanisms by which Pur-α suppresses neurotoxicity are still not clear. One hypothesis is that Pur-α binds and alters the G-quadruplex secondary structure of the G-rich repeats (Wortmann et al., 2020). In the context of FUS-ALS, Pur-α knockdown improves locomotor activity in mutant FUS flies, while Pur-α overexpression exacerbates FUS-ALS neurotoxicity (Di Salvio et al., 2015). In this context, a role of Pur-α in the regulation of stress granules and translation has been suggested by independent groups (Di Salvio et al., 2015; Daigle et al., 2016). In the context of Alzheimer’s disease, overexpression of Pur-α via stereotactic microinjection of AAV9-Pur-α significantly reduced Aβ deposition and pro-inflammatory cytokines in the hippocampus of APP/PS1 mice (Song et al. 2022).
So far, ~100 variants in PURA have been reported as pathogenic in the context of PURA syndrome. Of these, 25% are missense variants, 10% are 3-AA in-frame deletions, while the remaining 65% are protein-truncating variants (PTVs) caused by in-frame deletions, nonsense, and frameshift variants (Johannesen et al., 2021). The variants are distributed throughout the gene: 30% are located in the PUR-I repeat, 20% in the PUR-II repeat, and 25% in the PUR-III repeat, whereas the remainder are outside the PUR repeats. Nineteen variants were recurrent - most of them detected only in 2 or 3 patients, with p(Phe233del) being the most commonly reported variant in 14 patients (Johannesen et al., 2021).
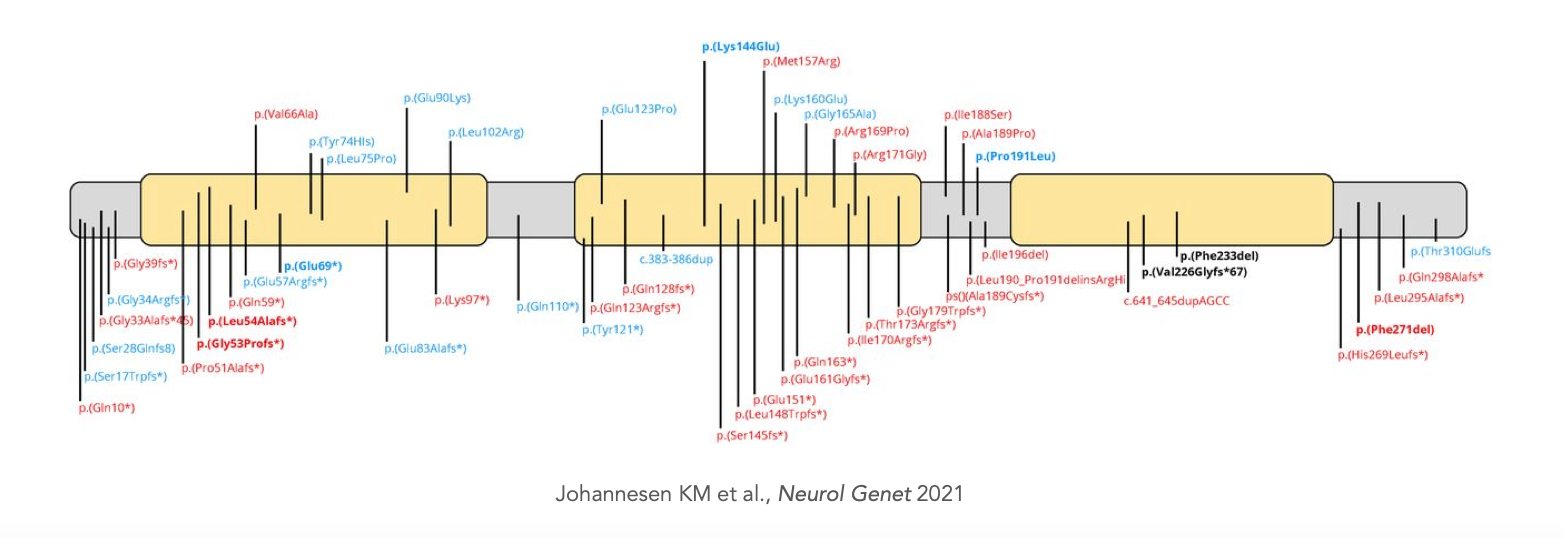
It has been suggested that disruption of PUR repeat III possibly results in a more severe phenotype (Hunt et al., 2014); however, genotype-phenotype correlation analyses have been inconclusive so far, and even within patients with identical variants, phenotype and severity can vary significantly (Cinquina et al., 2021; Johannesen et al., 2021). Given the apparent absence of mutational hotspots and genotype-phenotype correlation, haploinsufficiency due to loss of function has been assumed as the underlying mechanism of disease to explain the dominant phenotype.
While PURA syndrome phenotypes are heterogeneous, most patients do not achieve independent walking and remain nonverbal. The neonatal period is often complicated by respiratory insufficiency and feeding difficulties because of pronounced hypotonia. Treatment typically includes speech and language support as well as physical and occupational therapy and surgery to correct any orthopedic problems.
Despite the fact that PURA syndrome is known to cause severe neonatal hypotonia, only two recent studies (on a total number of 4 patients) have reported findings from muscle biopsies and electromyography. These findings are suggestive of a neuromuscular contribution to the symptoms (Mroczek et al., 2021; Wyrebek et al., 2022). Importantly, Wyrebek et al., 2022 showed a potential therapeutic role of pyridostigmine for PURA-related hypotonia. Pyridostigmine is an acetylcholinesterase inhibitor often used to treat neuromuscular junction disorders such as congenital myasthenic syndromes (Finsterer, 2019).
Currently, no therapeutics exist that restore cognitive or motor function.
Therapeutic Readiness
The development of new medicines requires disease models, patient-derived biological samples and cells, and a deep mechanistic understanding of pathophysiology that identifies disease modifying drug targets, including the monogenic driver gene itself.
Given that the pathophysiology of PURA syndrome and PURA mechanism(s) of action are not well understood, we believe that it is necessary to first seek to answer some basic mechanistic and functional questions before we can proceed with therapeutic approaches. Some of the limitations to our present understanding of PURA syndrome include:
Mechanism of disease
While ~100 PURA pathogenic variants have been reported, no functional studies have been performed to examine how these variants cause the disease. Haploinsufficiency due to loss of function has been assumed as the mechanism underlying the disease based on the similarity of phenotypes between PURA syndrome and 5q31 microdeletion, as well as the absence of a clear genotype-phenotype correlation comparing variants spread across the entire gene. However, it is important to note that the 5q31 microdeletions can encompass other genes such as neuregulin 2 (NRG2), a major gene for neurodevelopment (Brown et al., 2013). Moreover, the genotype-phenotype correlation studies might have been inconclusive because of the small size of the patient population.
Protein-truncating variants represent the majority of the PURA syndrome cases identified so far. PURA is a single-exon gene (i.e., lacks introns in its coding sequence), so while frameshift variants – like Jack’s c.533dupC (p.G179Wfs*22) variant – introduce premature stop codons, the mRNA is predicted to escape nonsense mediated mRNA decay (Maquat, 2004). Therefore, the translated protein product is likely made and may exhibit a dominant-negative effect or a gain-of-function mechanism.
It is imperative to characterize Jack’s and other PURA variants by studying the differences between the healthy vs the mutated versions of the protein. The first step will be to verify if the mutant protein is produced, and at what level compared to the wild type protein in the same patient’s cells and in healthy cells (isogenic control). If the mutant protein is produced, the next steps will be to compare the stability of the mutant protein to the wild-type protein, as well as to investigate its intracellular localization and functional properties. Importantly, it has been shown that Pur-α represses its own promoter activity and the region located between AA 1-85 is sufficient and necessary for this autoregulation (Muralidharan et al., 2001). If a truncated protein including this region is produced and stable, it might still be functional in this feedback mechanism.
Alignment of wild-type and Jack’s variant protein sequences

Critical functions and downstream targets
PURA has been suggested to be involved in the regulation of multiple processes including DNA replication and cell cycle progression, DNA repair, transcription, RNA transport, translation and membraneless organelle dynamics (stress granules, P-bodies and paraspeckles) (Di Salvio et al., 2015; Daigle et al., 2016; Molitor et al., 2021; Molitor et al., 2022). Therefore, Pur-α is likely affecting hundreds if not thousands of different downstream targets involved in multiple pathways. While it has not been determined which function(s) and target(s) of Pur-α are critical in the context of PURA syndrome, we propose to initially focus on the role of Pur-α in stress granule dynamics and control of mRNA transport and translation, and on a subset of downstream targets whose dysregulation might be consistent with the neurodevelopmental and neuromuscular phenotype of PURA patients.
Multiple independent studies have suggested a role of Pur-α in stress granule dynamics and in other neuronal RNP granules, where Pur-α co-localizes with other neuronal RNA-binding proteins, such as Pur-β, Staufen, hnRNPU, FMRP and a mutated FUS protein in the context of ALS (Di Salvio et al., 2015; Daigle et al., 2016; Li et al., 2001; Ohashi et al., 2000; Ohashi et al., 2002; Kanai et al., 2004; Mitsumori et al., 2017; Markmiller et al., 2018). Knockdown of endogenous Pur-α significantly reduced formation of cytoplasmic stress granules in mammalian cells suggesting that Pur-α is essential for the formation of stress granules (Daigle et al., 2016). The established methods for detecting stress granules provide ready tools for high-throughput assay development; therefore, this pathway might provide a screenable phenotype to optimize for a cell-based drug repurposing screen (Wang et al., 2020).
In neuronal cells, there is convincing evidence that Pur-α functions in mRNA transport. Pur-α associates with kinesin KIF5 and myosin Myo5a which both mediate RNA transport along neurites (Ohashi et al., 2002; Kanai et al., 2004), and has been suggested to regulate the transport of the dendritic protein MAP2 mRNA (Johnson et al., 2006). In neurons, Pur-α-positive granules are localized in both the shafts and spines of dendrites. Upon mGluR5 activation, Pur-α positive granules are transported in a Myo5a dependent manner into dendritic spines, where they colocalize with the postsynaptic factor PSD-95 (Mitsumori et al., 2017). Interestingly, in Pura knockout mice, the dendritic MAP2 protein is significantly reduced in whole brain lysates and mislocalized to the soma of knockout neurons and Purkinje cells (Hokkanen et al., 2012), likely because of impaired transport of its mRNA in absence of functional Pur-α (Johnson et al., 2006).
Other possible Pur-α downstream targets to prioritize are related to its transcriptional regulatory activity: myelin basic protein MBP in oligodendrocytes (Haas S et al., 1995), BC1 RNA in neural cells (Kobayashi et al., 2000), and MHC in skeletal muscle (Gupta et al., 2003; Ji et al., 2007; Pandey et al., 2020).
PURA has been implicated in the pathogenesis of other neurodevelopmental and neurological diseases, such as Fragile X-associated tremor/ataxia syndrome (FXTAS), Fragile X syndrome (FXS), two familial forms of Amyotrophic Lateral Sclerosis (C9orf72-ALS and FUS-ALS), and Alzheimer’s disease. Therefore, disease models and disease-modifying therapies developed for Jack’s Tomorrow may have broader applicability and impact across these and other related disorders.
PURA gene regulation
Currently, little is known about the transcriptional and post-transcriptional regulation of the PURA gene in different tissues and cell types or temporally during developmental, neonatal and postnatal/adult stages. We propose to couple basic research studies of transcriptional and post-transcriptional regulation with the design and development of ASOs for mutation-agnostic strategies to upregulate PURA expression (see the paragraph on therapeutic track/ASOs for a review on PURA gene regulation).
Dosage sensitivity, phenotype reversibility and therapeutic window of intervention
For a subset of genes in our genome, a change in gene dosage, by duplication or deletion, causes a phenotypic effect and therefore a strict control of protein levels is important for proper function. Overexpression of Pur-α is toxic in zebrafish (Penberthy et al., 2004) and flies (Di Salvio et al., 2015). No overexpression data is currently available on rodents except for one study involving localized overexpression of Pur-α in the hippocampus (Song et al., 2022). In addition, rescue experiments of Pura knockout mice – e.g., via gene replacement or transgenic expression – have never been performed; therefore, we have no knowledge regarding dosage sensitivity, phenotype reversibility, and potential therapeutic window of intervention.
Compensation by paralogs
Compensation of Purb and Purg in Pura knockout mice has not been explored. Simultaneous inactivation of Pura, Purb and Purg genes and combination rescue experiments in mice, along with the identification of common targets of Pur-α , Pur-β and Pur-γ might reveal overlapping or redundant functions of Pur family members in the future. It would be important to evaluate the expression of all paralogs in control and patient cells, to check if variants in Pur-α also affect paralog expression. How variants in Pur-α might affect heterodimerization with Pur-β and Pur-γ, and if these interactions are relevant to the pathogenesis of PURA syndrome is unknown.
Disease models
Cell-based and animal models of PURA syndrome will be useful for a range of downstream applications. These uses include: (i) further dissecting disease pathogenesis and pathophysiology in specific cell types or at specific developmental timepoints; (ii) biomarker discovery using transcriptomics, proteomics, and metabolomics; (iii) disease modifier and genetic suppressor screens; (iv) high-throughput drug repurposing and drug discovery screens.
Below is a description of the status of PURA disease models:
Cell models
To date there have been no peer reviewed reports of human stem cell disease modeling for PURA syndrome. We propose generating and characterizing three different truncation variant cell lines in the PUR-I, PUR-II (Jack’s variant) and PUR-III domains. We propose to generate iPSCs from fibroblasts and differentiate them and their isogenic control into neurons and skeletal muscle cells. In addition, recent protocols have been developed to create neuromuscular junctions in a dish via 3D co-culture of iPSC-derived muscle progenitors and motor neurons (Afshar Bakooshli et al., 2019; Pereira et al., 2021). These in vitro models will allow the characterization of PURA syndrome phenotypes and the establishment of functional assays for ASO and drug repurposing screens.
A primary phenotypic screen using patient derived skin fibroblasts will be established as a less expensive alternative to iPSCs. For example, a stress granule phenotype in fibroblasts would be compatible with an image-based high content screen and could be optimized for a cell-based drug repurposing screen (Wang et al., 2020).
Mouse models
Pur-α is highly conserved from bacteria to mammals with only a two amino acid difference between the human and murine protein. Two Pura knockout mouse models were previously generated by two independent groups and shown to recapitulate the main clinical phenotype of PURA patients (Khalili et al., 2003; Hokkanen et al., 2012; Barbe et al., 2016). However, both mouse models have limitations dictated by the strategy used for their generation and partially contradicting observations were reported. In particular, Khalili et al., 2003 reported decreased proliferation in the spleen, thymus, hippocampus, and cerebellum of Pura knockouts, while Hokkanen et al., 2012 reported megalencephaly with a significant enlargement of the stratum lacunosum-moleculare and higher proliferation rates in the hippocampus and cerebellum. Heterozygous mice display a less severe phenotype with still a significant reduction in number of neurons in the cerebellum and hippocampus, decreased tone, gait abnormalities, memory deficit and a higher prevalence of spontaneous early deaths (Barbe et al., 2016).
We propose to generate a new knockout mouse line in which Pura loss can be spatially restricted to specific organs or cellular lineages via the Cre-loxP system (conditional knockout), in order to dissect the cell autonomous and non-cell autonomous contributions of Pur-α to disease. In addition, we propose to generate patient avatar knockin mice to model PURA syndrome in vivo. The establishment of these mouse models will also be crucial to assess other critical gaps in the PURA syndrome knowledge base such as dosage sensitivity, phenotype reversibility and therapeutic window of intervention (e.g. via gene replacement or transgenic overexpression) and to test therapeutic approaches validated in human cells and invertebrate models.
Zebrafish
Pur-α sequence is highly conserved between human and the two Pur-α proteins in zebrafish (puraa 87% coverage, 80% identity; purab 85% coverage, 73% identity). The zebrafish Pur-α mostly differs from the human protein for the lack of a long N-term glycine stretch. This stretch is also absent in C. elegans.
In zebrafish, morpholinos against pura cause gata2 expression pooling at the animal pole and arrest in development at the gastrula stage (Penberthy et al., 2004). Data from morpholino knockdown should always be considered provisional and possibly artifactual until confirmed by a bona fide genetic model. A bona fide pura zebrafish model has not been published. Zebrafish has two pura genes, puraa and purab; therefore, both paralogs should be targeted for knockout.
Overexpression of Pur-α by injection of its mRNA causes a loss of gata2/GFP gene expression early in the ventral ectoderm and later in the nervous system, along with general developmental defects, and failure of the embryos to develop beyond 30 hpf (Penberthy et al., 2004). We do not recommend using zebrafish for modeling PURA syndrome patient variants given the presence of two pura genes.
Flies (Drosophila melanogaster)
Whereas humans have four known family members of the Pur family, Drosophila has only one Pur protein (73% coverage, 38% identity to human Pur-α). Di Salvio et al., 2015 have shown that in vivo downregulation of Pur-alpha via RNAi in all neurons or in motor neurons causes locomotion defects in Drosophila. In addition, in vivo overexpression of Pur-alpha in Drosophila neurons is also detrimental, and causes mild eye degeneration and unextended wings (Di Salvio et al., 2015). When overexpressed, Pur-alpha mitigates toxicities associated with Fragile X tumor ataxia syndrome (FXTAS) and C9orf72 repeat expansion diseases in Drosophila (Jin et al., 2007; Xu et al., 2013). However, in the context of FUS-ALS, Pur-alpha knockdown improves fly climbing activity of mutant FUS flies, while Pur-alpha overexpression exacerbates FUS-ALS neurotoxicity (Di Salvio et al., 2015).
Performing tissue-specific manipulations and generating transgenics is fast and cost-effective in Drosophila. Therefore, this model is ideal to address some critical questions like the cell-autonomous vs non-cell autonomous roles of Pur-alpha and to generate patient avatars to model specific variants. Drosophila is also amenable to high-throughput drug and genetic suppressor screens that could shed light on the critical pathways affected by Pur-α.
RNAi fly lines targeting Pur-alpha expression are available from the Bloomington Drosophila Stock Center at Indiana University Bloomington. An HA-Pur-alpha transgenic fly line (Di Salvio et al., 2015) is available from Dr. Gianluca Cestra’s lab (Italian National Research Council, Rome).
Worms (Caenorhabditis elegans)
Similar to fruit fly, worms can be used to quickly generate patient avatars for modeling specific variants and are also amenable to high-throughput drug and genetic suppressor screens.
The Pur-α orholog in Caenorhabditis elegans, Plp1 (66% coverage; 26% identity to human Pur-α), localizes to P granules throughout the germline and to similar perinuclear granules in somatic cells.
The homozygous plp-1 (ok2155) mutants display a temperature-sensitive sterility phenotype. Oocytes are absent in the plp-1 mutant, suggesting a requirement of plp-1 for germline differentiation (Lalani et al., 2014; Vishnupriya et al., 2020). The plp-1 mutants also have defective locomotion, marked by a 3-fold reduction in speed in comparison to wild-type animals (Lalani et al., 2014). It has been suggested that these defects are related to a role of Plp1 in mRNA trafficking during oogenesis and neuronal activities (Lalani et al., 2014).
RB1711 plp1(ok2155)IV worms are available from the Caenorhabditis Genetics Center at University of Minnesota.
Therapeutic Tracks
Track 1: ASOs
We will design variant specific ASOs to bypass the mutant allele (based on the outcome of Jack’s truncation protein validation in cells) and/or ASOs targeting various regulatory mechanisms to increase the levels of wild-type Pur-α. The goal is to rid the cell of mutated Pur-α (if needed), and to provide a functional level of healthy Pur-α. While transcriptional and post-transcriptional regulation of PURA requires additional study, we have identified potential regulatory mechanisms to target in order to up-regulate PURA expression:

Long non-coding RNA MALINC1
is located upstream to PURA in a head-to-head orientation and represses in cis expression of PURA. MALINC1 and PURA are both regulated by E2F1 (Bida et al., 2015; Darbinian et al., 2006). MALINC1 behaves as an oncogenic and immune-related lncRNA (Fabre et al., 2022); therefore potential side effects of its downregulation should be carefully evaluated.
Differently spliced 5’UTRs generated by three alternative promoters and TSS
PURA is under the complex regulation of three promoters driving expression of the same coding sequence from three different transcriptional start sites: the TSS I promoter is subject to feedback regulation by the Pur-α protein (Muralidharan et al., 2001); the TSS II promoter is downregulated through the presence of adjacent consensus binding elements for interferon regulatory factors (IRFs) and the intron from TSS II is removed more frequently in the brain than other tissues; the TSS III promoter is upregulated by E2F1, CAAT and NF-Y binding elements (Darbinian et al., 2006). It is not clear why PURA is under control of three different promoters and what is the evolutionary advantage of this regulation – a possibility is, for example, that non-coding RNAs derived from introns in the 5’-UTRs regulate mRNA translation (Darbinian et al., 2006).
MicroRNA binding sites in 3’UTRs
Translation of PURA has been shown to be inhibited by binding of microRNAs (miRNAs) to its 3’UTR in monocytes, resulting in modulation of the differentiation-dependent susceptibility of monocytic cells to HIV-1 infection (Shen et al., 2012). We propose to identify miRNA binding sites that might regulate PURA translation in neuronal and skeletal muscle cells. miRNA activity can potentially be blocked by complementary ASOs or RNAs that directly bind miRNAs (anti-miRNAs or miRNA sponges), or by ASOs that compete for their binding sites (miRNA masks). These strategies may result in increased protein expression (Ebert et al., 2007).
Potential alternative polyadenylation 3’UTR
Two principal isoforms of PURA (APPRIS P1: ENST00000331327.5 and ENST00000651386.1) are annotated in Ensembl, that differ from each other for having respectively a very long 3’UTR (10,496 bp) and a short UTR’(404 bp), possibly due to alternative polyadenylation. The different 3’UTRs may confer different transcript stability and/or translational efficiency and might be differentially regulated across cell types and tissues during development. However, this has not been investigated so far. ASOs could be potentially used to force changes in polyadenylation and increase production or one or the other isoform to upregulate Pur-α.
We recommend carrying out whole genome sequencing on Jack’s fibroblasts to provide the ability to design allele-specific ASOs.

Track 2: Drug repurposing
Drug repurposing is an attractive option for multiple reasons. First of all, FDA-approved drugs won’t need to be de-risked for use in humans to the same degree as novel compounds and this will significantly cut development timelines. Additionally, many of these drugs are produced commercially and are available at a fraction of the costs of a novel compound.
The Broad Repurposing Hub (commercially available as the SPECS library) is an ideal launchpad for identifying drug repurposing and drug repositioning candidates. The library includes 4,707 compounds, including 3,422 drugs that are currently marketed or have been tested in clinical trials in humans (Corsello et al., 2017). Drugs that have been approved or are currently in clinical trials for disorders related or similar to PURA syndrome and that are not included in the library will be added to the screen. These disorders include: congenital myasthenic syndromes, Fragile X-associated tremor/ataxia syndrome, Fragile X syndrome, Amyotrophic lateral sclerosis, Rett syndrome, Angelman syndrome, STXBP1 encephalopathy and SLC6A1 neurodevelopmental disorder, epilepsy, myoclonic epilepsies, proteinopathies, autism, disorders of myelination, leukodystrophy, Alzheimer’s and Parkinson’s disease. All validated PURA disease models will be screened against this collection as part of a coordinated multi-species high-throughput drug screening campaign.

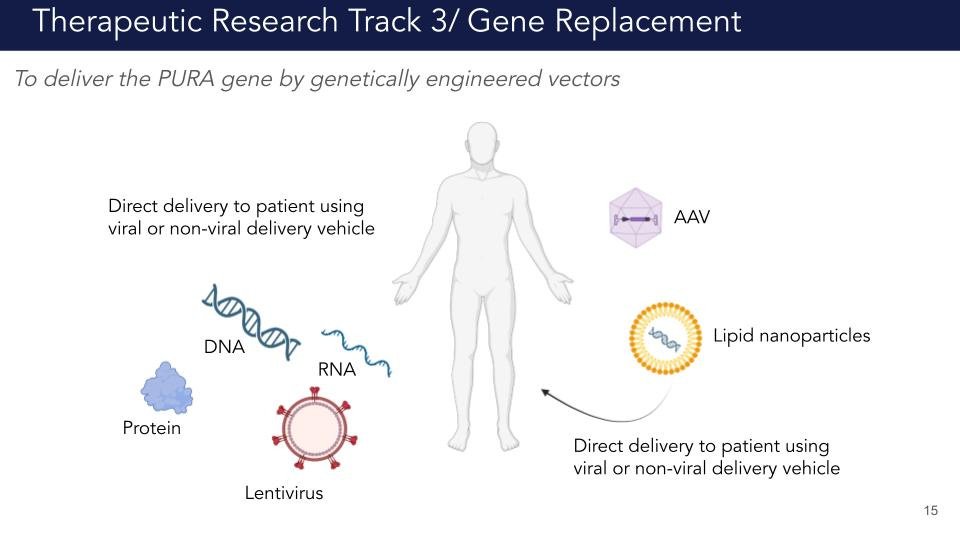
Track 3: Gene replacement
Two primary human PURA isoforms are annotated in the APPRIS and Ensembl databases: an 11.511kb isoform (ENST00000331327.5) that is too large for AAV packaging and a 1.919kb (ENST00000651386.1) that could be suitable for gene replacement. However, it is not known whether the two isoforms with 3’UTR of different lengths are differentially regulated, temporally or spatially. In addition, gene therapy approaches still face many challenges, such as viral delivery efficiency, immunogenicity and control of transgene expression. We plan to perform gene replacement proof of concept studies to test for efficacy and tolerability of PURA restoration in heterozygotes and knockout mice via AAV9. We will test AAV gene therapy at different neonatal and postnatal stages to test for phenotypic reversibility, dosage sensitivity and therapeutic window of intervention.
Additional therapeutic modalities (see Appendix)
DNA editing, RNA editing, protein replacement and new small molecule approaches need additional de-risking and therefore are not considered in Year One activities. These modalities will be revisited contingent on the outcome of ASO, drug repurposing and gene replacement tracks.
Join Jack’s Tomorrow in the fight!
References
Afshar Bakooshli M, Lippmann ES, Mulcahy B, Iyer N, Nguyen CT, Tung K, Stewart BA, van den Dorpel H, Fuehrmann T, Shoichet M, Bigot A, Pegoraro E, Ahn H, Ginsberg H, Zhen M, Ashton RS, Gilbert PM. A 3D culture model of innervated human skeletal muscle enables studies of the adult neuromuscular junction. Elife. 2019 May 14;8:e44530. doi: 10.7554/eLife.44530. PMID: 31084710; PMCID: PMC6516829.
Barbe MF, Krueger JJ, Loomis R, Otte J, Gordon J. Memory deficits, gait ataxia and neuronal loss in the hippocampus and cerebellum in mice that are heterozygous for Pur-alpha. Neuroscience. 2016 Nov 19;337:177-190. doi: 10.1016/j.neuroscience.2016.09.018. Epub 2016 Sep 17. PMID: 27651147; PMCID: PMC5458736.
Bergemann AD, Ma ZW, Johnson EM. Sequence of cDNA comprising the human pur gene and sequence-specific single-stranded-DNA-binding properties of the encoded protein. Mol Cell Biol. 1992a Dec;12(12):5673-82. doi: 10.1128/mcb.12.12.5673-5682.1992. PMID: 1448097; PMCID: PMC360507.
Bergemann AD, Johnson EM. The HeLa Pur factor binds single-stranded DNA at a specific element conserved in gene flanking regions and origins of DNA replication. Mol Cell Biol. 1992b Mar;12(3):1257-65. doi: 10.1128/mcb.12.3.1257-1265.1992. PMID: 1545807; PMCID: PMC369558.
Bida O, Gidoni M, Ideses D, Efroni S, Ginsberg D. A novel mitosis-associated lncRNA, MA-linc1, is required for cell cycle progression and sensitizes cancer cells to Paclitaxel. Oncotarget. 2015 Sep 29;6(29):27880-90. doi: 10.18632/oncotarget.4944. PMID: 26337085; PMCID: PMC4695032.
Brown N, Burgess T, Forbes R, McGillivray G, Kornberg A, Mandelstam S, Stark Z. 5q31.3 Microdeletion syndrome: clinical and molecular characterization of two further cases. Am J Med Genet A. 2013 Oct;161A(10):2604-8. doi: 10.1002/ajmg.a.36108. Epub 2013 Aug 15. PMID: 23950017.
Cinquina V, Ciaccio C, Venturini M, Masson R, Ritelli M, Colombi M. Expanding the PURA syndrome phenotype: A child with the recurrent PURA p.(Phe233del) pathogenic variant showing similarities with cutis laxa. Mol Genet Genomic Med. 2021 Jan;9(1):e1562. doi: 10.1002/mgg3.1562. Epub 2020 Dec 4. PMID: 33275834; PMCID: PMC7963414.
Cooper-Knock J, Walsh MJ, Higginbottom A, Robin Highley J, Dickman MJ, Edbauer D, Ince PG, Wharton SB, Wilson SA, Kirby J, Hautbergue GM, Shaw PJ. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain. 2014 Jul;137(Pt 7):2040-51. doi: 10.1093/brain/awu120. Epub 2014 May 27. PMID: 24866055; PMCID: PMC4065024.
Corsello SM, Bittker JA, Liu Z, Gould J, McCarren P, Hirschman JE, Johnston SE, Vrcic A, Wong B, Khan M, Asiedu J, Narayan R, Mader CC, Subramanian A, Golub TR. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat Med. 2017 Apr 7;23(4):405-408. doi: 10.1038/nm.4306. PMID: 28388612; PMCID: PMC5568558.
Daigle JG, Krishnamurthy K, Ramesh N, Casci I, Monaghan J, McAvoy K, Godfrey EW, Daniel DC, Johnson EM, Monahan Z, Shewmaker F, Pasinelli P, Pandey UB. Pur-α regulates cytoplasmic stress granule dynamics and ameliorates FUS toxicity. Acta Neuropathol. 2016 Apr;131(4):605-20. doi: 10.1007/s00401-015-1530-0. Epub 2016 Jan 4. PMID: 26728149; PMCID: PMC4791193.
Daniel DC, Johnson EM. PURA, the gene encoding Pur-alpha, member of an ancient nucleic acid-binding protein family with mammalian neurological functions. Gene. 2018 Feb 15;643:133-143. doi: 10.1016/j.gene.2017.12.004. Epub 2017 Dec 6. PMID: 29221753; PMCID: PMC5770235.
Darbinian N, White MK, Khalili K. Regulation of the Pur-alpha promoter by E2F-1. J Cell Biochem. 2006 Nov 1;99(4):1052-63. doi: 10.1002/jcb.20872. PMID: 16741925.
Di Salvio M, Piccinni V, Gerbino V, Mantoni F, Camerini S, Lenzi J, Rosa A, Chellini L, Loreni F, Carrì MT, Bozzoni I, Cozzolino M, Cestra G. Pur-α functionally interacts with FUS carrying ALS-associated mutations. Cell Death Dis. 2015 Oct 22;6(10):e1943. doi: 10.1038/cddis.2015.295. PMID: 26492376; PMCID: PMC4632316.
Finsterer J. Congenital myasthenic syndromes. Orphanet J Rare Dis. 2019 Feb 26;14(1):57. doi: 10.1186/s13023-019-1025-5. PMID: 30808424; PMCID: PMC6390566.
Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007 Sep;4(9):721-6. doi: 10.1038/nmeth1079. Epub 2007 Aug 12. PMID: 17694064; PMCID: PMC3857099.
Fabre ML, Canzoneri R, Gurruchaga A, Lee J, Tatineni P, Kil H, Lacunza E, Aldaz CM, Abba MC. MALINC1 an Immune-Related Long Non-Coding RNA Associated with Early-Stage Breast Cancer Progression. Cancers (Basel). 2022 Jun 7;14(12):2819. doi: 10.3390/cancers14122819. PMID: 35740485; PMCID: PMC9221538.
Gallia GL, Johnson EM, Khalili K. Puralpha: a multifunctional single-stranded DNA- and RNA-binding protein. Nucleic Acids Res. 2000 Sep 1;28(17):3197-205. doi: 10.1093/nar/28.17.3197. PMID: 10954586; PMCID: PMC110688.
Graebsch A, Roche S, Niessing D. X-ray structure of Pur-alpha reveals a Whirly-like fold and an unusual nucleic-acid binding surface. Proc Natl Acad Sci U S A. 2009 Nov 3;106(44):18521-6. doi: 10.1073/pnas.0907990106. Epub 2009 Oct 21. PMID: 19846792; PMCID: PMC2765457.
Gupta M, Sueblinvong V, Raman J, Jeevanandam V, Gupta MP. Single-stranded DNA-binding proteins PURalpha and PURbeta bind to a purine-rich negative regulatory element of the alpha-myosin heavy chain gene and control transcriptional and translational regulation of the gene expression. Implications in the repression of alpha-myosin heavy chain during heart failure. J Biol Chem. 2003 Nov 7;278(45):44935-48. doi: 10.1074/jbc.M307696200. Epub 2003 Aug 21. PMID: 12933792.
Haas S, Thatikunta P, Steplewski A, Johnson EM, Khalili K, Amini S. A 39-kD DNA-binding protein from mouse brain stimulates transcription of myelin basic protein gene in oligodendrocytic cells. J Cell Biol. 1995 Sep;130(5):1171-9. doi: 10.1083/jcb.130.5.1171. PMID: 7657701; PMCID: PMC2120554.
Hokkanen S, Feldmann HM, Ding H, Jung CK, Bojarski L, Renner-Müller I, Schüller U, Kretzschmar H, Wolf E, Herms J. Lack of Pur-alpha alters postnatal brain development and causes megalencephaly. Hum Mol Genet. 2012 Feb 1;21(3):473-84. doi: 10.1093/hmg/ddr476. Epub 2011 Oct 18. PMID: 22010047.
Hosoki K, Ohta T, Natsume J, Imai S, Okumura A, Matsui T, Harada N, Bacino CA, Scaglia F, Jones JY, Niikawa N, Saitoh S. Clinical phenotype and candidate genes for the 5q31.3 microdeletion syndrome. Am J Med Genet A. 2012 Aug;158A(8):1891-6. doi: 10.1002/ajmg.a.35439. Epub 2012 Jun 18. PMID: 22711443.
Hunt D, Leventer RJ, Simons C, Taft R, Swoboda KJ, Gawne-Cain M; DDD study, Magee AC, Turnpenny PD, Baralle D. Whole exome sequencing in family trios reveals de novo mutations in PURA as a cause of severe neurodevelopmental delay and learning disability. J Med Genet. 2014 Dec;51(12):806-13. doi: 10.1136/jmedgenet-2014-102798. Epub 2014 Oct 23. PMID: 25342064; PMCID: PMC4251168.
Ji J, Tsika GL, Rindt H, Schreiber KL, McCarthy JJ, Kelm RJ Jr, Tsika R. Puralpha and Purbeta collaborate with Sp3 to negatively regulate beta-myosin heavy chain gene expression during skeletal muscle inactivity. Mol Cell Biol. 2007 Feb;27(4):1531-43. doi: 10.1128/MCB.00629-06. Epub 2006 Dec 4. PMID: 17145772; PMCID: PMC1800711.
Jin P, Duan R, Qurashi A, Qin Y, Tian D, Rosser TC, Liu H, Feng Y, Warren ST. Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron. 2007 Aug 16;55(4):556-64. doi: 10.1016/j.neuron.2007.07.020. PMID: 17698009; PMCID: PMC1994817.
Johannesen KM, Gardella E, Gjerulfsen CE, Bayat A, Rouhl RPW, Reijnders M, Whalen S, Keren B, Buratti J, Courtin T, et al. PURA-Related Developmental and Epileptic Encephalopathy: Phenotypic and Genotypic Spectrum. Neurol Genet. 2021 Nov 15;7(6):e613. doi: 10.1212/NXG.0000000000000613. PMID: 34790866; PMCID: PMC8592566.
Johnson EM, Kinoshita Y, Weinreb DB, Wortman MJ, Simon R, Khalili K, Winckler B, Gordon J. Role of Pur alpha in targeting mRNA to sites of translation in hippocampal neuronal dendrites. J Neurosci Res. 2006 May 1;83(6):929-43. doi: 10.1002/jnr.20806. PMID: 16511857.
Johnson EM, Daniel DC, Gordon J. The pur protein family: genetic and structural features in development and disease. J Cell Physiol. 2013 May;228(5):930-7. doi: 10.1002/jcp.24237. PMID: 23018800; PMCID: PMC3747735.
Kanai Y, Dohmae N, Hirokawa N. Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron. 2004 Aug 19;43(4):513-25. doi: 10.1016/j.neuron.2004.07.022. PMID: 15312650.
Kariolis MS, Wells RC, Getz JA, Kwan W, Mahon CS, Tong R, Kim DJ, Srivastava A, Bedard C, Henne KR, Giese T, Assimon VA, Chen X, Zhang Y, Solanoy H, Jenkins K, Sanchez PE, Kane L, Miyamoto T, Chew KS, Pizzo ME, Liang N, Calvert MEK, DeVos SL, Baskaran S, Hall S, Sweeney ZK, Thorne RG, Watts RJ, Dennis MS, Silverman AP, Zuchero YJY. Brain delivery of therapeutic proteins using an Fc fragment blood-brain barrier transport vehicle in mice and monkeys. Sci Transl Med. 2020 May 27;12(545):eaay1359. doi: 10.1126/scitranslmed.aay1359. PMID: 32461332.
Kelm RJ Jr, Elder PK, Strauch AR, Getz MJ. Sequence of cDNAs encoding components of vascular actin single-stranded DNA-binding factor 2 establish identity to Puralpha and Purbeta. J Biol Chem. 1997 Oct 17;272(42):26727-33. doi: 10.1074/jbc.272.42.26727. PMID: 9334258.
Khalili K, Del Valle L, Muralidharan V, Gault WJ, Darbinian N, Otte J, Meier E, Johnson EM, Daniel DC, Kinoshita Y, Amini S, Gordon J. Puralpha is essential for postnatal brain development and developmentally coupled cellular proliferation as revealed by genetic inactivation in the mouse. Mol Cell Biol. 2003 Oct;23(19):6857-75. doi: 10.1128/MCB.23.19.6857-6875.2003. PMID: 12972605; PMCID: PMC193944.
Kobayashi S, Agui K, Kamo S, Li Y, Anzai K. Neural BC1 RNA associates with pur alpha, a single-stranded DNA and RNA binding protein, which is involved in the transcription of the BC1 RNA gene. Biochem Biophys Res Commun. 2000 Oct 22;277(2):341-7. doi: 10.1006/bbrc.2000.3683. PMID: 11032728.
Lalani SR, Zhang J, Schaaf CP, Brown CW, Magoulas P, Tsai AC, El-Gharbawy A, Wierenga KJ, Bartholomew D, et al.,Mutations in PURA cause profound neonatal hypotonia, seizures, and encephalopathy in 5q31.3 microdeletion syndrome. Am J Hum Genet. 2014 Nov 6;95(5):579-83. doi: 10.1016/j.ajhg.2014.09.014. Epub 2014 Oct 16. PMID: 25439098; PMCID: PMC4225583.
Li Y, Koike K, Ohashi S, Funakoshi T, Tadano M, Kobayashi S, Anzai K, Shibata N, Kobayashi M. Pur alpha protein implicated in dendritic RNA transport interacts with ribosomes in neuronal cytoplasm. Biol Pharm Bull. 2001 Mar;24(3):231-5. doi: 10.1248/bpb.24.231. PMID: 11256476.
Liu H, Johnson EM. Distinct proteins encoded by alternative transcripts of the PURG gene, located contrapodal to WRN on chromosome 8, determined by differential termination/polyadenylation. Nucleic Acids Res. 2002 Jun 1;30(11):2417-26. doi: 10.1093/nar/30.11.2417. PMID: 12034829; PMCID: PMC117198.
Maquat LE. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol. 2004 Feb;5(2):89-99. doi: 10.1038/nrm1310. PMID: 15040442.
Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, Luo EC, Krach F, Yang D, Sen A, Fulzele A, Wozniak JM, Gonzalez DJ, Kankel MW, Gao FB, Bennett EJ, Lécuyer E, Yeo GW. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell. 2018 Jan 25;172(3):590-604.e13. doi: 10.1016/j.cell.2017.12.032. PMID: 29373831; PMCID: PMC5969999.
Mitsumori K, Takei Y, Hirokawa N. Components of RNA granules affect their localization and dynamics in neuronal dendrites. Mol Biol Cell. 2017 Jun 1;28(11):1412-1417. doi: 10.1091/mbc.E16-07-0497. Epub 2017 Apr 12. PMID: 28404748; PMCID: PMC5449141.
Molitor L, Bacher S, Burczyk S, Niessing D. The Molecular Function of PURA and Its Implications in Neurological Diseases. Front Genet. 2021 Mar 11;12:638217. doi: 10.3389/fgene.2021.638217. PMID: 33777106; PMCID: PMC7990775.
Molitor L, Klostermann M, Bacher S, Spranger N, Merl-Pham J, Rusha E, Tews D, Pertek A, Proske M, Busch A, Reschke S, Burczyk S, Feederle R, Hauck S, Blum H, Drukker M, Fischer-Posovsky P, König J, Zarnack K, Niessing D. Depletion of the RNA-binding protein PURA triggers changes in posttranscriptional gene regulation and loss of P-bodies. bioRxiv 2022.02.09.479353; doi: https://doi.org/10.1101/2022.02.09.479353.
Mroczek M, Zafeiriou D, Gurgel-Gianetti J, Vilela Morais de Azevedo B, Roos A, Bartels E, Kohlschmidt N, Phadke R, Feng L, Duff J, Töpf A, Straub V. Three Individuals with PURA Syndrome in a Cohort of Patients with Neuromuscular Disease. Neuropediatrics. 2021 Oct;52(5):390-393. doi: 10.1055/s-0040-1715625. Epub 2020 Dec 22. PMID: 33352606.
Muralidharan V, Sweet T, Nadraga Y, Amini S, Khalili K. Regulation of Puralpha gene transcription: evidence for autoregulation of Puralpha promoter. J Cell Physiol. 2001 Mar;186(3):406-13. doi: 10.1002/1097-4652(2000)9999:999<000::AID-JCP1039>3.0.CO;2-P. PMID: 11169980.
Ohashi S, Kobayashi S, Omori A, Ohara S, Omae A, Muramatsu T, Li Y, Anzai K. The single-stranded DNA- and RNA-binding proteins pur alpha and pur beta link BC1 RNA to microtubules through binding to the dendrite-targeting RNA motifs. J Neurochem. 2000 Nov;75(5):1781-90. doi: 10.1046/j.1471-4159.2000.0751781.x. PMID: 11032866.
Ohashi S, Koike K, Omori A, Ichinose S, Ohara S, Kobayashi S, Sato TA, Anzai K. Identification of mRNA/protein (mRNP) complexes containing Puralpha, mStaufen, fragile X protein, and myosin Va and their association with rough endoplasmic reticulum equipped with a kinesin motor. J Biol Chem. 2002 Oct 4;277(40):37804-10. doi: 10.1074/jbc.M203608200. Epub 2002 Jul 29. PMID: 12147688.
Pandey PR, Yang JH, Tsitsipatis D, Panda AC, Noh JH, Kim KM, Munk R, Nicholson T, Hanniford D, Argibay D, Yang X, Martindale JL, Chang MW, Jones SW, Hernando E, Sen P, De S, Abdelmohsen K, Gorospe M. circSamd4 represses myogenic transcriptional activity of PUR proteins. Nucleic Acids Res. 2020 Apr 17;48(7):3789-3805. doi: 10.1093/nar/gkaa035. PMID: 31980816; PMCID: PMC7144931.
Penberthy WT, Zhao C, Zhang Y, Jessen JR, Yang Z, Bricaud O, Collazo A, Meng A, Lin S. Pur alpha and Sp8 as opposing regulators of neural gata2 expression. Dev Biol. 2004 Nov 1;275(1):225-34. doi: 10.1016/j.ydbio.2004.08.007. PMID: 15464585.
Pereira JD, DuBreuil DM, Devlin AC, Held A, Sapir Y, Berezovski E, Hawrot J, Dorfman K, Chander V, Wainger BJ. Human sensorimotor organoids derived from healthy and amyotrophic lateral sclerosis stem cells form neuromuscular junctions. Nat Commun. 2021 Aug 6;12(1):4744. doi: 10.1038/s41467-021-24776-4. PMID: 34362895; PMCID: PMC8346474.
Reijnders MRF, Janowski R, Alvi M, Self JE, van Essen TJ, Vreeburg M, Rouhl RPW, Stevens SJC, Stegmann APA, Schieving J,et al.. PURA syndrome: clinical delineation and genotype-phenotype study in 32 individuals with review of published literature. J Med Genet. 2018 Feb;55(2):104-113. doi: 10.1136/jmedgenet-2017-104946. Epub 2017 Nov 2. PMID: 29097605; PMCID: PMC5800346.
Ring HZ, Chang H, Guilbot A, Brice A, LeGuern E, Francke U. The human neuregulin-2 (NRG2) gene: cloning, mapping and evaluation as a candidate for the autosomal recessive form of Charcot-Marie-Tooth disease linked to 5q. Hum Genet. 1999 Apr;104(4):326-32. doi: 10.1007/s004390050961. PMID: 10369162.
Rossi S, Serrano A, Gerbino V, Giorgi A, Di Francesco L, Nencini M, Bozzo F, Schininà ME, Bagni C, Cestra G, Carrì MT, Achsel T, Cozzolino M. Nuclear accumulation of mRNAs underlies G4C2-repeat-induced translational repression in a cellular model of C9orf72 ALS. J Cell Sci. 2015 May 1;128(9):1787-99. doi: 10.1242/jcs.165332. Epub 2015 Mar 18. PMID: 25788698.
Shen CJ, Jia YH, Tian RR, Ding M, Zhang C, Wang JH. Translation of Pur-α is targeted by cellular miRNAs to modulate the differentiation-dependent susceptibility of monocytes to HIV-1 infection. FASEB J. 2012 Nov;26(11):4755-64. doi: 10.1096/fj.12-209023. Epub 2012 Jul 26. PMID: 22835829.
Scholefield J, Harrison PT. Prime editing - an update on the field. Gene Ther. 2021 Aug;28(7-8):396-401. doi: 10.1038/s41434-021-00263-9. Epub 2021 May 24. PMID: 34031549; PMCID: PMC8376635.
Shimojima K, Isidor B, Le Caignec C, Kondo A, Sakata S, Ohno K, Yamamoto T. A new microdeletion syndrome of 5q31.3 characterized by severe developmental delays, distinctive facial features, and delayed myelination. Am J Med Genet A. 2011 Apr;155A(4):732-6. doi: 10.1002/ajmg.a.33891. Epub 2011 Mar 15. Erratum in: Am J Med Genet A. 2011 Nov;155A(11):2903. PMID: 21594995.
Song C, Zhang Y, Huang W, Shi J, Huang Q, Jiang M, Qiu Y, Wang T, Chen H, Wang H. Circular RNA Cwc27 contributes to Alzheimer's disease pathogenesis by repressing Pur-α activity. Cell Death Differ. 2022 Feb;29(2):393-406. doi: 10.1038/s41418-021-00865-1. Epub 2021 Sep 9. PMID: 34504314; PMCID: PMC8817017.
Steinauer A, LaRochelle JR, Knox SL, Wissner RF, Berry S, Schepartz A. HOPS-dependent endosomal fusion required for efficient cytosolic delivery of therapeutic peptides and small proteins. Proc Natl Acad Sci U S A. 2019 Jan 8;116(2):512-521. doi: 10.1073/pnas.1812044116. PMID: 30610181; PMCID: PMC6329960.
Swinnen B, Bento-Abreu A, Gendron TF, Boeynaems S, Bogaert E, Nuyts R, Timmers M, Scheveneels W, Hersmus N, Wang J, Mizielinska S, Isaacs AM, Petrucelli L, Lemmens R, Van Damme P, Van Den Bosch L, Robberecht W. A zebrafish model for C9orf72 ALS reveals RNA toxicity as a pathogenic mechanism. Acta Neuropathol. 2018 Mar;135(3):427-443. doi: 10.1007/s00401-017-1796-5. Epub 2018 Jan 4. PMID: 29302778.
Tanaka AJ, Bai R, Cho MT, Anyane-Yeboa K, Ahimaz P, Wilson AL, Kendall F, Hay B, Moss T, Nardini M, Bauer M, Retterer K, Juusola J, Chung WK. De novo mutations in PURA are associated with hypotonia and developmental delay. Cold Spring Harb Mol Case Stud. 2015 Oct;1(1):a000356. doi: 10.1101/mcs.a000356. PMID: 27148565; PMCID: PMC4850890.
Vishnupriya R, Thomas L, Wahba L, Fire A, Subramaniam K. PLP-1 is essential for germ cell development and germline gene silencing in Caenorhabditis elegans. Development. 2020 Nov 27;147(22):dev195578. doi: 10.1242/dev.195578. PMID: 33051256; PMCID: PMC7710023.
Wang F, Li J, Fan S, Jin Z, Huang C. Targeting stress granules: A novel therapeutic strategy for human diseases. Pharmacol Res. 2020 Nov;161:105143. doi: 10.1016/j.phrs.2020.105143. Epub 2020 Aug 16. PMID: 32814168; PMCID: PMC7428673.
Weber J, Bao H, Hartlmüller C, Wang Z, Windhager A, Janowski R, Madl T, Jin P, Niessing D. Structural basis of nucleic-acid recognition and double-strand unwinding by the essential neuronal protein Pur-alpha. Elife. 2016 Jan 8;5:e11297. doi: 10.7554/eLife.11297. PMID: 26744780; PMCID: PMC4764581.
White MK, Johnson EM, Khalili K. Multiple roles for Puralpha in cellular and viral regulation. Cell Cycle. 2009 Feb 1;8(3):1-7. doi: 10.4161/cc.8.3.7585. Epub 2009 Feb 10. PMID: 19182532; PMCID: PMC2683411.
Wortman MJ, Dagdanova AV, Clark AM, Godfrey EW, Pascal SM, Johnson EM, Daniel DC. A synthetic Pur-based peptide binds and alters G-quadruplex secondary structure present in the expanded RNA repeat of C9orf72 ALS/FTD. Biochim Biophys Acta Mol Cell Res. 2020 Jun;1867(6):118674. doi: 10.1016/j.bbamcr.2020.118674. Epub 2020 Feb 6. PMID: 32035967; PMCID: PMC7323587.
Wyrebek R, DiBartolomeo M, Brooks S, Geller T, Crenshaw M, Iyadurai S. Hypotonic infant with PURA syndrome-related channelopathy successfully treated with pyridostigmine. Neuromuscul Disord. 2022 Feb;32(2):166-169. doi: 10.1016/j.nmd.2022.01.005. Epub 2022 Jan 16. PMID: 35094889.
Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, Li H, Hales CM, Gearing M, Wingo TS, Jin P. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A. 2013 May 7;110(19):7778-83. doi: 10.1073/pnas.1219643110. Epub 2013 Apr 3. PMID: 23553836; PMCID: PMC3651485.
Zhang X, Zoltek M, Mozumdar D, Schepartz A. Dose-dependent nuclear delivery and transcriptional repression with a cell-penetrant MeCP2. bioRxiv 2022.06.03.494754; doi: https://doi.org/10.1101/2022.06.03.494754
Appendix
Additional therapeutic modalities
Therapeutic Track 4: Prime editing
Prime editing is a relatively novel method of genome editing that can perform all possible nucleotide substitutions and resolve frameshifts induced by indels (like in Jack’s variant) with lower off-target probability when compared with conventional CRISPR/Cas systems (Scholefield et al., 2021). This approach has the advantage of fixing sequence errors without overexpressing. However, the treatment is irreversible, and the application of genome editing approaches for targeting neurodevelopmental monogenetic disorders is still in its infancy and will require a longer path to approval. Companies like Prime Medicine could be engaged for interest in a preclinical proof-of-concept study to edit DNA via prime editing in patient cells.
Therapeutic Track 5: RNA editing or stop codon readthrough
An RNA editing strategy could be developed for PURA point mutations, and a suppressor tRNA strategy could be developed for premature stop codon readthrough to bypass premature stop codons in frameshift mutations. By targeting RNA – and not DNA – these approaches are not irreversible. However, the downside is that they might require redosing, in addition to the same delivery challenges of gene replacement and prime editing (delivery efficiency and immunogenicity). Companies like Shape Therapeutics could be engaged for interest in a preclinical proof-of-concept study to edit or readthrough RNA in patient cells.
Therapeutic Track 6: Protein replacement
Cell-permeant miniature proteins are currently being investigated in the lab of Dr. Alanna Schepartz at UC Berkeley as a strategy for delivering functional Mecp2 protein in Rett syndrome. This approach exploits a pathway for endosomal release that involves the HOPS complex, a natural endosome remodeling machine (Steinauer et al., 2019). Proof of concept studies have shown efficient dose-dependent delivery of a functional cell-penetrant Mecp2 in cells (Zhang et al., 2022). Protein replacement would make it possible to titrate the dose of protein allowing to provide the right amount to improve symptoms without side effects of overexpression. Since proteins are subject to degradation, this therapeutic would need to be provided on an ongoing basis. The lab of Dr. Alanna Schepartz will be engaged for interest in a preclinical proof-of-concept study to test the applicability of this approach for Pur-α. The biggest challenge with this strategy would be delivering sufficient amounts of protein to the target tissues in vivo across. As an alternative approach, Denali Therapeutics could be engaged for interest in sharing their platform based on engineered transferrin receptor to deliver protein therapeutics across the BBB to the brain (Kariolis et al., 2020).
Therapeutic Track 7: Novel small molecule discovery
Along with repurposing already-approved drugs, it is conceivable to develop a new chemical entity with therapeutic utility in patients with PURA syndrome. A scaled-up functional drug screen based on the previous drug repurposing track including drugs that may not be FDA approved might be limited and quite costly. In the scenario of a dominant negative or gain of function mechanism of disease, it would be possible to consider direct binders/inhibitors of mutant Pur-α. If the truncation variants are expressed and assembled in the multimeric complex it would also be possible to design an in vitro screen (structurally oriented) to identify compounds able to interfere specifically with mutant protein recruitment (also by drug repurposing).

YOUR SUPPORT MATTERS
Your support directly helps us fund important work like this.
Consider a donation today and join the Jack’s Tomorrow team as we find and fund a cure for PURA Syndrome.